Research
研究内容
4. 表皮におけるセラミドとバリア形成
表皮は基底層,有棘層,顆粒層,角質層の四層から構成されており,最外層の角質層が主に皮膚の透過性のバリア(皮膚バリア)を形成している(図 9)。皮膚バリアは感染症,炎症,アレルギー疾患の原因となる病原体,異物,アレルゲンの侵入を防ぎ,体内からの水分の損失を抑えるという重要な役割をもつ。そのため,バリア機能の低下は感染症,アトピー性皮膚炎,魚鱗癬,乾皮症などの皮膚疾患のリスク増大あるいは原因となる。
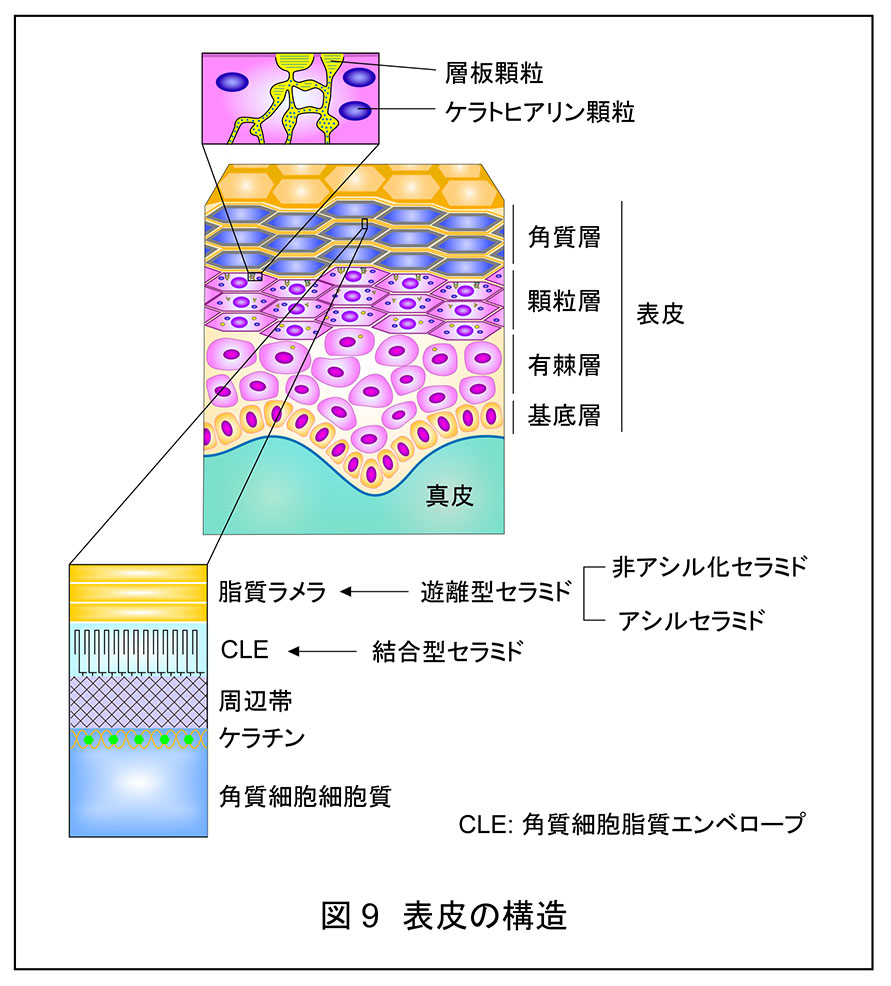
皮膚バリア形成で重要な役割を果たす角質層の構造体のうち,セラミドは脂質ラメラと角質細胞脂質エンベロープ(CLE)の構成成分である。脂質ラメラは角質細胞間に存在する脂質の多層構造体であり,ヒトには 13 nm の長周期層と 6 nm の短周期層の層が見られる。このうち 13 nm の長周期層が生物種を問わず共通して見られることから皮膚バリアに重要であると推測されている。脂質ラメラにはセラミド,コレステロール,遊離脂肪酸がおおよそ等モルずつ存在すると考えられている。セラミドはタンパク質と共有結合していない遊離型と共有結合した結合型に大別されるが,脂質ラメラを構成するセラミドは遊離型である。CLE は角質細胞の表面を覆う細胞膜様の構造体であり,周辺帯タンパク質と結合した結合型セラミドにより構成される。周辺帯はタンパク質の架橋構造体であり,角質細胞に物理的な強度を与えている。角質細胞は外界と接しているため様々な環境変化に対応しなくてはならない。そのため,角質細胞は通常の脂質二重膜のような脆弱な膜構造ではなく,CLE のような共有結合性の強固な脂質構造体を必要としたのであろう。角質層に存在するセラミドは主に顆粒層で産生される。顆粒層で産生されたセラミドは一旦グルコシル化された状態(グルコシルセラミド;一部はホスホコリンが付加されたスフィンゴミエリン)で層板顆粒に蓄えられる。層板顆粒は顆粒と名前がついているが,最近の研究からチューブ状のネットワーク構造であると考えられる。層板顆粒に蓄えられたグルコシルセラミド/スフィンゴミエリンは顆粒層と角質層の境界部で細胞外へ放出され,極性基が除去されてセラミドに戻る。
セラミドは長鎖塩基と脂肪酸がアミド結合した構造をもつ。ヒトには 5 種類の長鎖塩基 [スフィンゴシン (S), ジヒドロスフィンゴシン (DS), フィトスフィンゴシン (P), 6-ヒドロキシスフィンゴシン (H), 4,14-スフィンガジエン(SD)] と 5 種類の脂肪酸 [非水酸化脂肪酸 (N), α-水酸化脂肪酸 (A), ω-水酸化脂肪酸 (O),エステル化 ω-水酸化脂肪酸 (EO),タンパク質結合脂肪酸 (P-O/P-EO)] の組み合わせからなる 25 クラスのセラミドが存在し,それぞれのクラスは脂肪酸と長鎖塩基の略号の組み合わせで表記される(例:NS,EOS など; 図 10A, B)。EO をもつセラミドはアシルセラミド(正確にはω-O-アシルセラミド)と呼ばれ,エステル化された脂肪酸のほとんどはリノール酸である(図 10C)。遊離型セラミドのうち,アシルセラミド以外を非アシル化セラミドと呼ぶ。脂質ラメラを構成するセラミドのほとんどは非アシル化セラミドである。アシルセラミドは非アシル化セラミドに比べて量的には少ないが,その特徴的な構造によって脂質ラメラの多層構造を形成/維持する役割をもつため,皮膚バリア形成において重要である。アシルセラミドの一部は結合型セラミドへと変換される。その過程では,リノール酸部分が過酸化,エポキシ/水酸化,エポキシ/エノン化と連続的に修飾を受ける。結合型セラミドの構造に関して,修飾を受けたリノール酸がリパーゼによって除去されて,ω-水酸化セラミドとなった後,ω-水酸基部分でタンパク質のグルタミン酸残基とエステル結合するという構造モデル(図 10A;タンパク質結合ω-水酸化脂肪酸 [P-O] モデル)が長年信じられてきた。一方,2020 年にエポキシ/エノン化したアシルセラミドのエノン部分がタンパク質とマイケル付加反応あるいはシッフ塩基形成をしているという新たな構造モデル(図10A;タンパク質結合修飾リノール酸エステル化ω-水酸化脂肪酸 [P-EO] モデル)が提唱された。生化学研究室は,この P-EO モデルの結合型セラミドの構造決定と実際の表皮における存在の有無について検討を行った結果,マウス表皮において P-EO タイプの結合型セラミドの存在を確認し,それらがマイケル付加反応でシステイン残基に結合したものであることを明らかにした23)(プレスリリース原稿)。一方,少なくとも我々の測定条件下では従来から提唱されてきた P-O タイプの結合型セラミドおよびシステイン残基以外に結合した P-EO セラミドは検出されなかった。
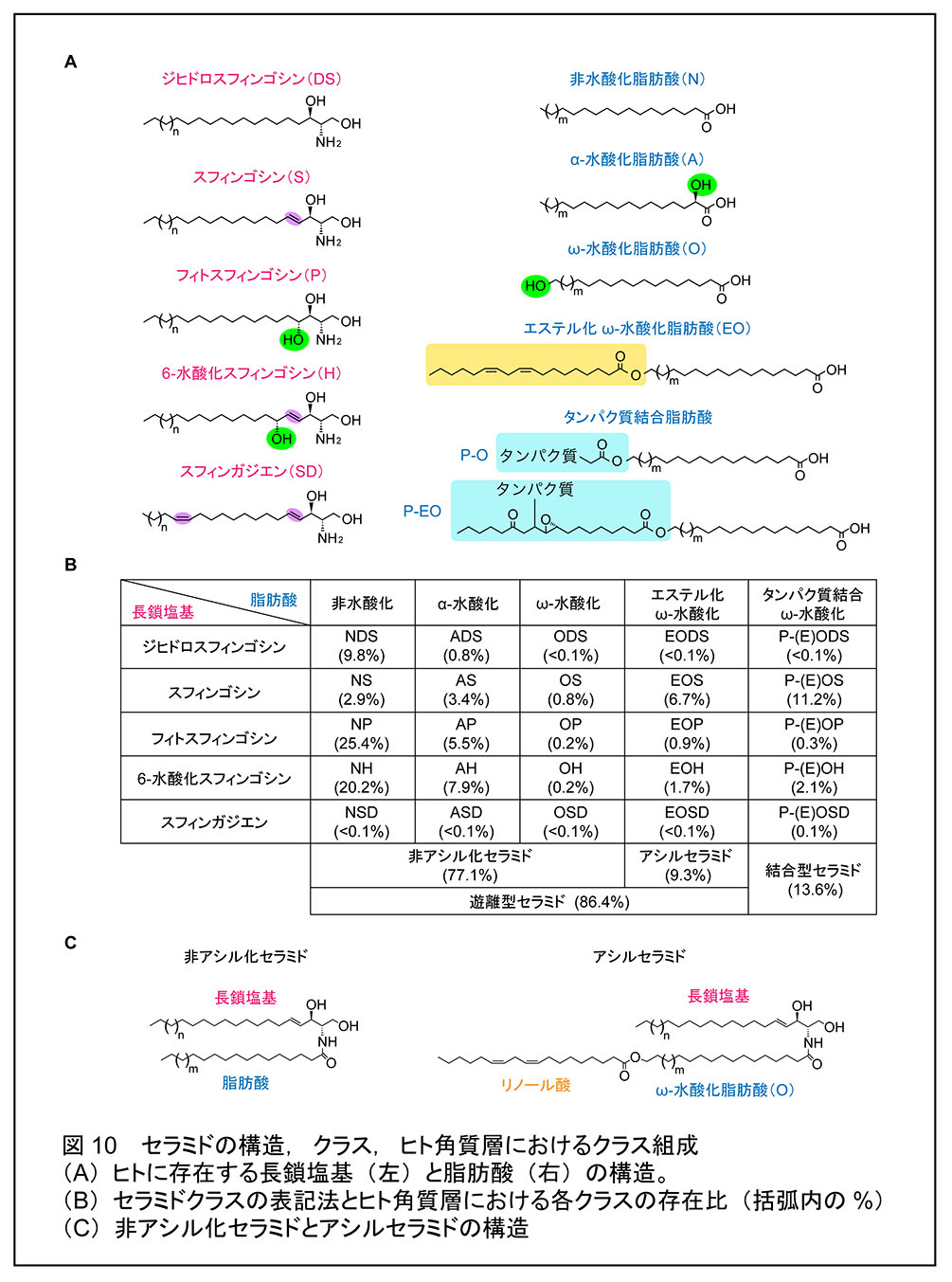
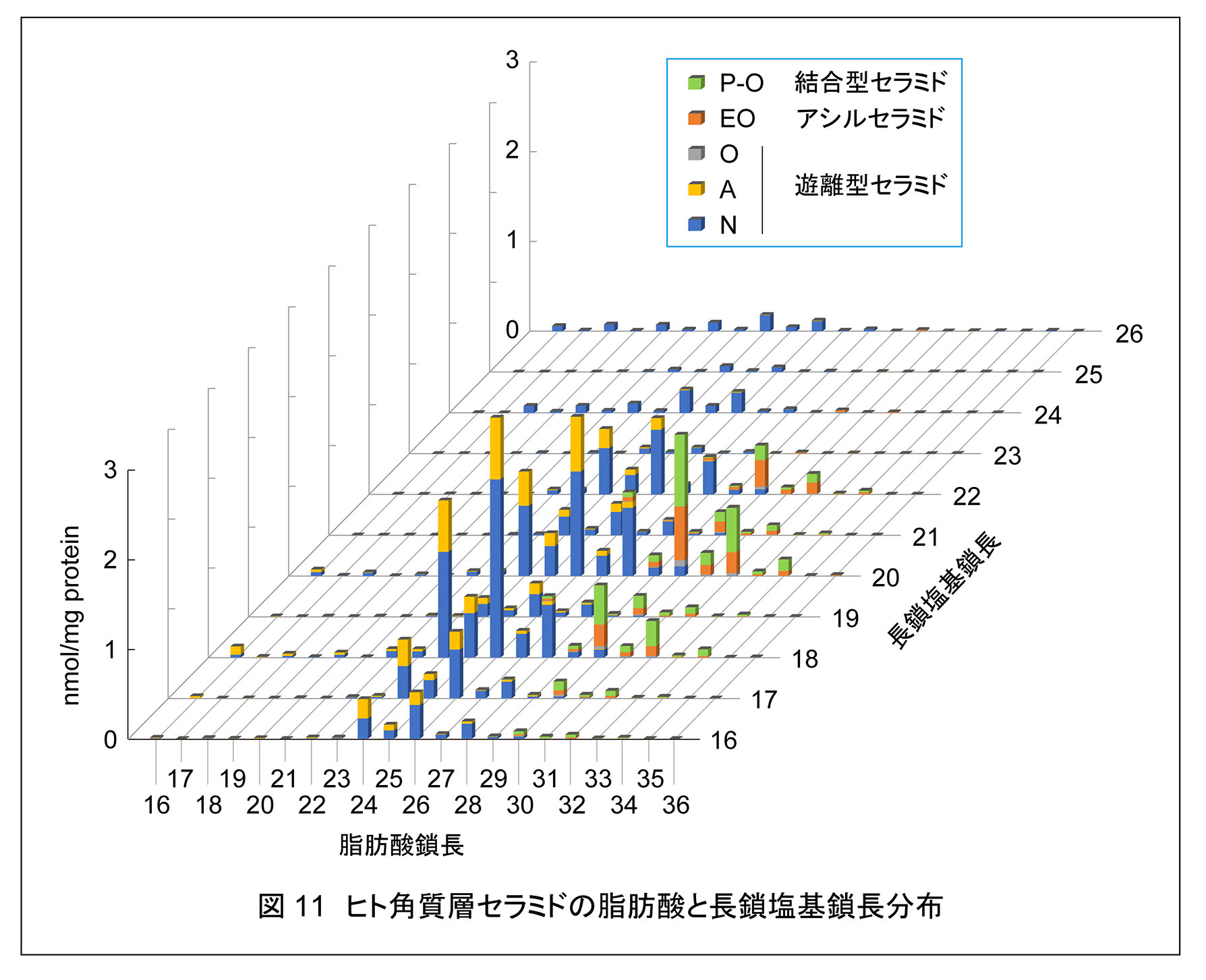
哺乳類の多くの組織のセラミド組成は比較的単純であり,存在するセラミドのクラスは NS,NDS,NSD のみである。このうち,NS が最も主要である。NSD は腎臓に多く存在する。また,NP は表皮,食道,小腸,大腸,腎臓などの上皮系の組織に存在し,AS は脳のミエリンに豊富に存在する。一方,表皮には多様かつ多量のセラミドが存在し,EO 含有セラミド(アシルセラミド)や H 含有セラミドは表皮にのみ存在する。それぞれのクラスには脂肪酸および長鎖塩基の鎖長(脂肪酸は不飽和度の違いも含む)が異なった複数のセラミド分子種が存在する。生化学研究室では表皮角質層のセラミドを分離・定量できる質量分析法(液体クロマトグラフィー連結タンデム質量分析法;LC-MS/MS)を確立し,2020 年にまず,長鎖塩基鎖長を C18 に絞った解析によってヒト角質層に 26 クラス/408 分子種,マウスに 21 クラス/342 分子種のセラミドが存在することを明らかにした1)。さらに 2022 年に,長鎖塩基鎖長を C16 から C26 に拡大した解析によって,ヒト角質層に 23 クラス/1581 分子種のセラミドが存在することを明らかにし,ヒト角質層のセラミドの全容を解明することに成功した17)(プレスリリース原稿)。後者の解析においてセラミドクラス数が減少しているのは,微量のセラミドをカウントしなかったためである。なお,この解析における結合型セラミドの測定では,結合型セラミドをアルカリ処理することによって O 型セラミド(P-O タイプと P-EO タイプの結合型セラミド両方に共通した成分)に変換して測定しているため,両者の区別はできていない。
一般的な組織におけるセラミドの脂肪酸鎖長は C16 から C24,長鎖塩基鎖長はほとんど C18 に限られるのに対し,ヒト角質層セラミド中の脂肪酸鎖長は C16 から C36,長鎖塩基鎖長は C16 から C26 と幅広い(図 11)。角質層セラミド中の脂肪酸の長さはクラスによって特徴が見られる。非アシル化セラミド中の N 型と A 型セラミドすなわち,非水酸化脂肪酸あるいはα-水酸化脂肪酸をもつセラミドの脂肪酸鎖長は C16 から C28 までであり,そのうち C24 と C26 が多い。一方,非アシル化セラミド中の O 型,アシルセラミド,結合型セラミドの脂肪酸(ω-水酸化脂肪酸)部分の鎖長は C28 から C36 であり,そのうち C30,C32,C34 が多い。一般的な組織と角質層のセラミドの脂肪酸鎖長の違いは,細胞膜の脂質二重層(10 nm)と角質層脂質ラメラ(長周期;13 nm)の厚みの違いを反映している。ヒト角質層に多様なセラミド分子種が存在する意義は環境変化に対応するためであろう。脂質ラメラはゲル相を形成しているが,環境変化(温度,pH,湿度,塩濃度など)によって相転移が起きないようになっていなければならない。脂質組成の不均一性は相転移温度幅を広げることができるため,角質層に多様なセラミド分子種が存在することによって脂質ラメラのゲル相が安定化されると考えられる。また,角質層中にコレステロール量が多いこともゲル相の安定化に寄与しているのであろう。
ヒトの角質層にはフィトスフィンゴシン(P)あるいは 6-水酸化スフィンゴシン(H)をもつ P 型,H 型セラミドが多いことが特徴である。総セラミド量に対する割合は NP が 25.4%,NH が 20.2%,AP が 5.5%,AH が 7.9% であり,これらの合計では約 6 割に達する(図 10B)。これらの長鎖塩基には NS などの一般的な組織に多いスフィンゴシンよりも1つ水酸基が多く存在し,この水酸基を介した水素結合が脂質ラメラにおける脂質ー脂質間相互作用を強めていると思われる。一方,長鎖塩基の中で唯一シス二重結合をもつ 4,14-スフィンガジエン(SD)を含む SD 型セラミドは角質層にはほとんど存在しない。その理由は 14 位のシス二重結合が脂質ラメラ中での脂質の密なパッキングを妨げるからであろう。アトピー性皮膚炎患者では,表皮のセラミドの量の低下,クラス組成の変化,脂肪酸の短鎖化が生じることが知られている。このうち,クラス組成の変化としては長鎖塩基側に水酸基の多い非アシル化セラミド NP,AP とアシルセラミド EOS,EOH,EOP の減少が見られる。
スフィンゴシンを含有する S 型セラミドおよびフィトスフィンゴシンを含有する P 型セラミド(フィトセラミド)はジヒドロスフィンゴシンをもつ DS 型セラミド(ジヒドロセラミド)のそれぞれ 4 位不飽和化あるいは 4 位水酸化によって生じる。これらを触媒する酵素はそれぞれ DEGS1 と DEGS2 である。DEGS2 は水酸化活性だけでなく,不飽和化活性ももつ二機能酵素である。生化学研究室では基質であるジヒドロセラミドの脂肪酸鎖長によってその活性の使い分けがあること,すなわち脂肪酸部分が極長鎖である場合には水酸化活性が優勢になることを見出した21)。DEGS2 が産生するフィトセラミド NP はヒト角質層において最も量が多いセラミドクラスであり,皮膚バリア機能に重要な役割を果たしていると思われる。一方,マウス表皮角質層における NP 量は少なく(全体の約 1 %),Degs2 KO マウスは皮膚バリア異常を示さない。
上述の通り,アシルセラミドは脂質ラメラの形成/維持,結合型セラミドは CLE 形成に関わるため,これらは皮膚バリア形成に極めて重要である。アシルセラミドおよび結合型セラミド形成に関わる遺伝子の多くは 2010 年以降に同定された。これらの遺伝子の変異はいずれもヒトでは先天性魚鱗癬を引き起こし,遺伝子 KO マウスは皮膚バリア異常で新生致死となる。生化学研究室ではアシルセラミド形成に関わる遺伝子の多くを同定し,その産生経路の詳細(反応の基質,順序,種類)について解明してきた。生化学研究室で明らかにしたアシルセラミド合成経路における6つの反応は下記と図 12 の通りである。
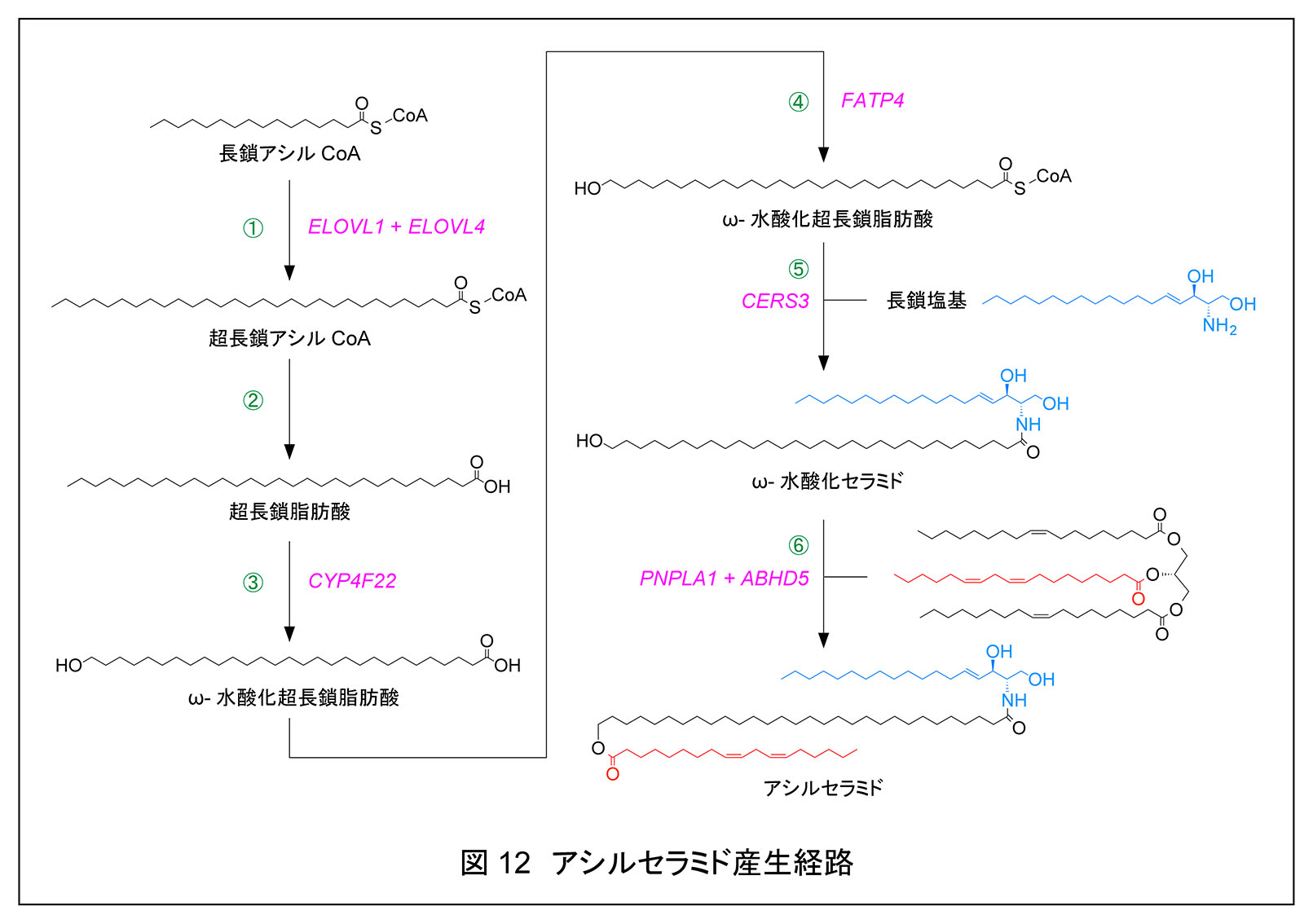
生化学研究室では上述の通り,Elovl1,Cyp4f39,Fatp4 の KO マウスを作成・解析し,いずれも新生致死性の皮膚バリア形成異常を示すことを見出したが2, 7, 8),これらの KO マウスの皮膚バリア機能異常の重篤度は Cyp4f39 KO,Elovl1 KO,Fatp4 KO マウスの順であり,この重篤度の違いはアシルセラミド量の減少の程度の差だけでなく,結合型セラミド量の減少や KO した遺伝子ごとに特徴的なセラミド組成変化も寄与していた24)。
生化学研究室では発症原因が不明な他の魚鱗癬原因遺伝子の解析も行ってきた。NIPAL4 は Mg2+ トランスポーターをコードするが,その変異が魚鱗癬を引き起こす分子機構は不明であった。生化学研究室では Nipal4 KO マウスを作成・解析し,その魚鱗癬様表現型がアシルセラミド(ω-O-アシルセラミド)の産生低下に起因することを明らかにした14)。また,Nipal4 KO マウスの表皮では 1-O-アシルセラミドという別のタイプのアシルセラミドの産生が誘導され,通常タイプのアシルセラミドであるω-O-アシルセラミドは量の低下に加えて O-アシル鎖の組成変化も引き起こされていることを見出した25)。野生型マウスではω-O-アシルセラミドの O-アシル鎖のほとんどはリノール酸であるのに対して(89%),KO マウスではその比率が 66% まで減少し,他の脂肪酸種が増加した(特にオレイン酸が 2% から 16% に上昇)。正常なケラチノイトでは分化に依存して細胞内 Mg2+ 濃度が上昇するのに対し,Nipal4 KO マウス由来ケラチノサイトではそのような上昇が見られなかった。Mg2+ にはリン脂質に結合して膜を安定化する作用がある。ω-O-アシルセラミドなどの特殊な脂質を産生する表皮顆粒層の小胞体は本来 NIPAL4 依存的な Mg2+ 濃度上昇によって安定化されていると思われるが,KO マウスではそのような Mg2+ 濃度上昇が起こらないために小胞体膜の物性が変化し,通常とは異なる脂質が産生されるようになった可能性がある。
脂肪族アルデヒドデヒドロゲナーゼ ALDH3A2 は魚鱗癬症候群であるシェーグレン・ラルソン症候群の原因遺伝子である。生化学研究室では ALDH3A2 が長鎖塩基の分解経路で生じる長鎖アルデヒドの代謝に関与することを明らかにしてきた(詳細は 「2.生理活性脂質スフィンゴシン1-リン酸の生理機能と代謝」を参照)。Aldh3a2 KO マウスはヒトでは偽遺伝子である Aldh3b2 との重複のため,通常条件下では皮膚バリア形成異常を示さなかった。ただし,角質層を障害した場合の皮膚バリアの回復には遅延が見られた15)。一方,Aldh3a2 Aldh3b2 二重 KO マウスは魚鱗癬様表現型を示し,その原因はアシルセラミド産生低下であった16)。このマウスでは蓄積した長鎖アルデヒドがアシルセラミド産生に働く酵素を攻撃することでアシルセラミド産生を低下させていると考えられる。アシルセラミド量の低下は,Aldh3a2 Aldh3b2 二重欠損マウスだけではなく,ALDH3A2 欠損ヒトケラチノサイト,シェーグレン・ラルソン症候群患者においても観察された19)。以上,直接的にはアシルセラミド産生には関わらない NIPAL4 や ALDH3A2 の変異によっても間接的にアシルセラミドの産生が低下することで魚鱗癬が引き起こされていることが明らかとなった。
生化学研究室ではこれらアシルセラミド産生の分子機構の解明を通じて,アシルセラミドの増加によるバリア機能改善の方策を見いだし,アトピー性皮膚炎を始めとする皮膚疾患の新たな治療につなげたいと考えている。また,生化学研究室では,アシルセラミド,結合型セラミドが表皮だけでなく,マウスの口腔から前胃の粘膜上皮にも存在し,口腔バリア形成に重要な役割を果たしていることを見出した22)(プレスリリース原稿)。これらの組織で脂肪酸伸長酵素遺伝子 Elovl1 が欠損したコンディショナル KO マウスでは,食道と舌の形態異常,蛍光色素の浸透亢進,カプサイシン飲料に対する忌避行動が観察された。また,ヒトにおいても,少なくとも解析した頬粘膜と歯肉にはアシルセラミドが存在し,歯肉には結合型セラミドもが存在していた。このように表皮だけと思われていた透過性バリア形成におけるセラミドの重要性は口腔(とおそらく消化管上部)にも拡大した。
研究業績
- Kawana M, Miyamoto M, Ohno Y, Kihara A (2020) Comparative profiling and comprehensive quantification of stratum corneum ceramides in humans and mice by LC-MS/MS. J Lipid Res, 61, 884-895.
- Sassa T, Ohno Y, Suzuki S, Nomura T, Nishioka C, Kashiwagi T, Hirayama T, Akiyama M, Taguchi R, Shimizu H, Itohara S, Kihara A (2013) Impaired epidermal permeability barrier in mice lacking the Elovl1 gene responsible for very long-chain fatty acid production. Mol Cell Biol, 33, 2787-2796.
- Ohno Y, Suto S, Yamanaka M, Mizutani Y, Mitsutake S, Igarashi Y, Sassa T, Kihara A (2010) ELOVL1 production of C24 acyl-CoAs is linked to C24 sphingolipid synthesis. Proc Natl Acad Sci USA, 107, 18439-18444.
- 4Mueller N, Sassa T, Morales-Gonzalez S, Schneider J, Salchow DJ, Seelow D, Knierim E, Stenzel W, Kihara A, Schuelke M (2019) De novo mutation in ELOVL1 causes ichthyosis, acanthosis nigricans, hypomyelination, spastic paraplegia, high frequency deafness and optic atrophy. J Med Genet, 56, 164-175.
- Ohno Y, Nakamichi S, Ohkuni A, Kamiyama N, Naoe A, Tsujimura H, Yokose U, Sugiura K, Ishikawa J, Akiyama M, and Kihara A (2015) Essential role of the cytochrome P450 CYP4F22 in the production of acylceramide, the key lipid for skin permeability barrier formation. Proc. Natl. Acad. Sci. USA, 112, 7707-7712.(プレスリリース原稿)
- Nohara T, Ohno Y, Kihara A. (2021) Impaired production of the skin barrier lipid acylceramide by CYP4F22 ichthyosis mutations. J Dermatol Sci, 101, 69-71.
- Miyamoto M, Itoh N, Sawai M, Sassa T, Kihara A (2020) Severe skin permeability barrier dysfunction in knockout mice deficient in a fatty acid ω-hydroxylase crucial to acylceramide production. J Invest Dermatol, 140, 319-326.
- Yamamoto H, Hattori M, Chamulitrat W, Ohno Y, Kihara A (2020) Skin permeability barrier formation by the ichthyosis-causative gene FATP4 through formation of the barrier lipid ω-O-acylceramide. Proc Natl Acad Sci USA, 117, 2914-2922.(プレスリリース原稿)
- Mizutani Y, Kihara A, Igarashi Y (2006) LASS3 (longevity assurance homologue 3) is a mainly testis-specific (dihydro)ceramide synthase with relatively broad substrate specificity. Biochem J, 398, 531-538.
- Mizutani Y, Kihara A, Chiba H, Tojo H, Igarashi Y (2008) 2-Hydroxy-ceramide synthesis by ceramide synthase family: enzymatic basis for the preference of FA chain length. J Lipid Res, 49, 2356-2364.
- Yamamoto M, Sassa T, Kyono Y, Uemura H, Kugo M, Hayashi H, Imai Y, Yamanishi K, Kihara A. (2021) Comprehensive stratum corneum ceramide profiling reveals reduced acylceramides in ichthyosis patient with CERS3 mutations. J Dermatol, 48, 447-456.
- Ohno Y, Kamiyama N, Nakamichi S, Kihara A. (2017) PNPLA1 is a transacylase essential for the generation of the skin barrier lipid ω-O-acylceramide. Nat Commun, 8, 14610.(プレスリリース原稿)
- Ohno Y, Nara A, Nakamichi S, Kihara A (2018) Molecular mechanism of the ichthyosis pathology of Chanarin-Dorfman syndrome: Stimulation of PNPLA1-catalyzed ω-O-acylceramide production by ABHD5. J Dermatol Sci, 92, 245-253.
- Honda Y, Kitamura T, Naganuma T, Abe T, Ohno Y, Sassa T, Kihara A. (2018) Decreased skin barrier lipid acylceramide and differentiation-dependent gene expression in ichthyosis gene Nipal4 knockout mice. J Invest Dermatol, 138, 741-749.
- Naganuma T, Takagi S, Kanetake T, Kitamura T, Hattori S, Miyakawa T, Sassa T, Kihara A. (2016) Disruption of the Sjögren-Larsson syndrome gene Aldh3a2 in mice increases keratinocyte growth and retards skin barrier recovery. J Biol Chem, 291,11676-11688.
- Nojiri K, Fudetani S, Arai A, Kitamura T, Sassa T, Kihara A. (2021) Impaired skin barrier function due to reduced ω-O-acylceramide levels in a mouse model of Sjögren-Larsson syndrome. Mol Cell Biol, 41, e0035221.
- Suzuki M, Ohno Y, Kihara A. (2022) Whole picture of human stratum corneum ceramides, including the chain-length diversity of long-chain bases. J Lipid Res, 63, 100235.(プレスリリース原稿)
(ASBMB (2022) vol.21, No. 10, p18で紹介) - Nohara T, Ohno Y, Kihara A. (2022) Impaired production of skin barrier lipid acylceramides and abnormal localization of PNPLA1 due to ichthyosis-causing mutations in PNPLA1. J Dermatol Sci, 107, 89-94.
- Arai A, Takeichi T, Wakamoto H, Sassa T, Ito Y, Murase Y, Ogi T, Akiyama M, Kihara A. (2022) Ceramide profiling of stratum corneum in Sjögren-Larsson syndrome. J Dermatol Sci, 107, 114-122.
- Takahashi T, Turker S, Sassa T, Akcapinar GB, Yararbas K, Susguna S, Iseri SAU, Kihara A, Akcakaya NH. (2022) Hypomyelinating spastic dyskinesia and ichthyosis caused by a homozygous splice site mutation leading to exon skipping in ELOVL1. Brain Dev, 44, 391-400.
- Ota A, Morita H, Naganuma T, Miyamoto M, Jojima K, Nojiri K, Matsuda J, Kihara A. (2023) Bifunctional DEGS2 has higher hydroxylase activity toward substrates with very-long-chain fatty acids in the production of phytosphingosine-ceramides. J Biol Chem, 104603.
- Sassa T, Kihara A. (2023) Involvement of ω-O-acylceramides and protein-bound ceramides in oral permeability barrier formation. Cell Rep, 42, 112363. (プレスリリース原稿)
- Ohno Y, Nakamura T, Iwasaki T, Katsuyama A, Ichikawa S, Kihara A. (2023) Determining the structure of protein-bound ceramides, essential lipids for skin barrier function. iScience, 26, 108248. (プレスリリース原稿)
- Yamamoto Y, Sassa T, Kihara A. (2024) Comparison of skin barrier abnormalities and epidermal ceramide profiles among three ω-O-acylceramide synthesis-deficient mouse strains. J Dermatol Sci, 113, 10-17.
- Yamaji M, Ohno Y, Shimada M, Kihara A. (2024) Alteration of epidermal lipid composition as a result of deficiency in the magnesium transporter Nipal4. J Lipid Res, 300, 105656.